
Pleiotropy
Statins Effects: What's The Real Story?

Over the years I have written quite a bit on the topic of cholesterol and statins. Today I will be coming form a slightly different angle. This is not meant to defend statins as a drug. It is meant to explain why some research shows benefit. Although not in mortality as much as softer end points which do matter and we will discuss. This is meant to add to the knowledge base and make us wonder if there are other ways to both get the benefit of what statins do seem to offer in some, while avoiding the side effects they produce in many. And remember that even if a side effect isn’t objectively measured, no pharmaceutical intervention has only one intended effect.
On with the show…
Statins & Mortality: No Clear Win in Primary Prevention
A landmark 2010 meta-analysis by Kausik Ray and colleagues, published in the Archives of Internal Medicine, pulled together 11 randomized controlled trials involving 65,229 high-risk participants with no prior cardiovascular disease. These were people with elevated risk factors—diabetes, hypertension, smoking—but no history of heart attack or stroke.
Key result: Statins produced no statistically significant reduction in all-cause mortality. The risk ratio was 0.91 (95% CI 0.83–1.01). In plain numbers, over roughly 244,000 person-years of follow-up, there were about 100 fewer deaths in the statin groups, but the confidence interval crossed 1.0, meaning it could easily have been zero benefit or even slight harm. The result held when diabetes-only trials were excluded and showed no evidence of publication bias. (1)
Crucially, there was no correlation between the degree of LDL-C reduction (average ~40 mg/dL drop) or baseline LDL levels and the mortality benefit. Age drove most of the variation in event rates, not cholesterol lowering. The authors concluded: “Statin therapy for an average period of 3.7 years had no benefit on all-cause mortality in a high-risk primary prevention population.”
Subsequent reviews have echoed variations of this. In low-risk primary prevention, absolute mortality benefits are tiny (often <0.5% absolute risk reduction over 5 years). Even in broader meta-analyses that include some higher-risk groups, all-cause mortality reductions are modest at best, and absolute benefits remain small when you account for the millions of people treated. Secondary prevention (post-event) shows clearer gains, but that’s a different population with active disease.
The pattern raises an obvious question: If LDL lowering is the primary mechanism, why doesn’t greater LDL reduction reliably translate into fewer total deaths? And why do the survival curves sometimes separate early—within months—before major atherosclerotic regression could plausibly occur?
Pleiotropic Effects: Real, but What’s the Star Player?
Statins do far more than just lower LDL cholesterol by blocking the enzyme HMG-CoA reductase in the liver. This blockade doesn’t stop at cholesterol production—it disrupts the entire mevalonate pathway, reducing the synthesis of intermediate molecules called isoprenoids (specifically farnesyl pyrophosphate [FPP] and geranylgeranyl pyrophosphate [GGPP]). These isoprenoids are essential for a process called prenylation, which attaches lipid anchors to certain proteins, allowing them to function properly in cell membranes and signaling.
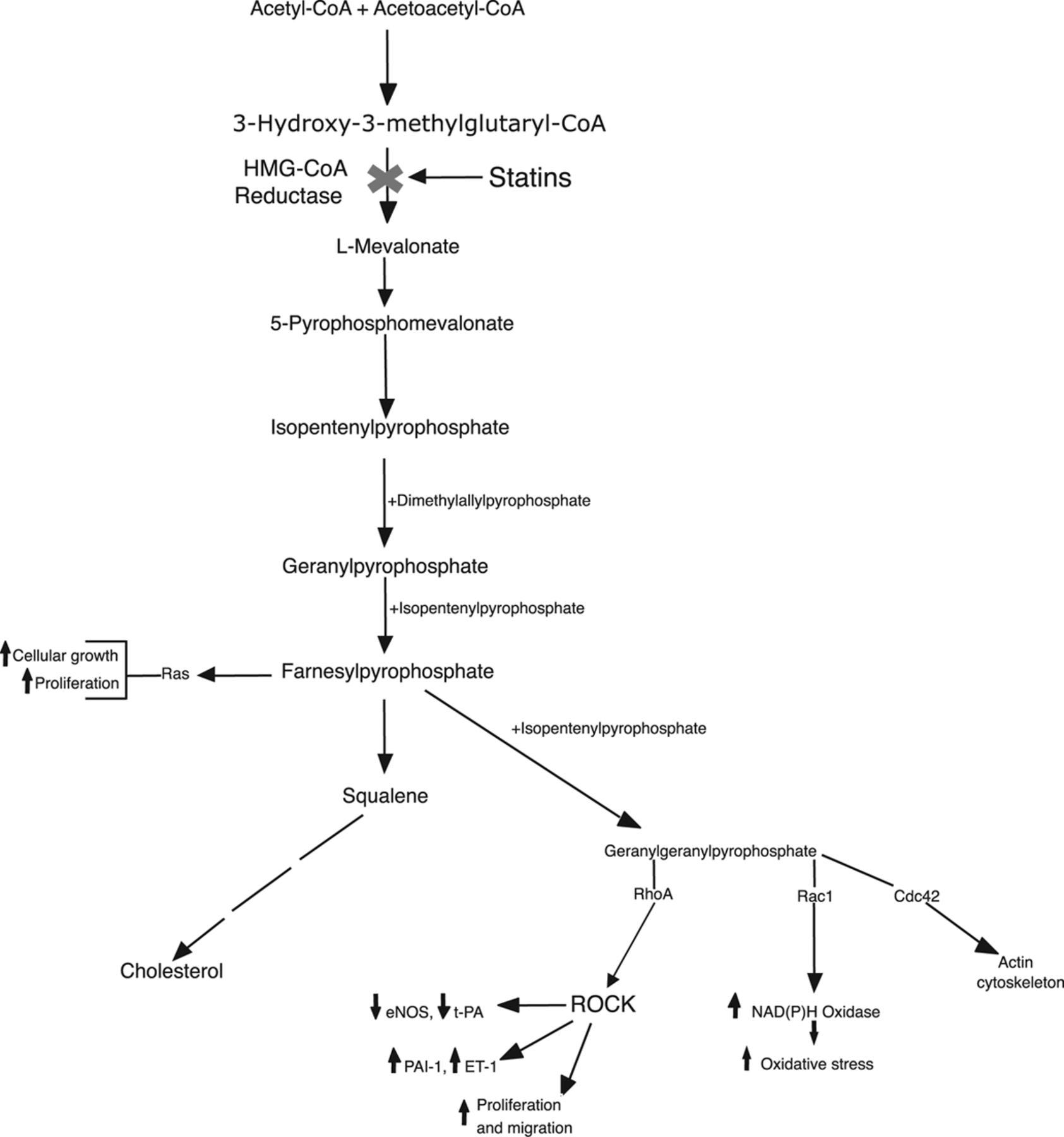
The key proteins affected are small GTPases like Rho, Rac, and Ras. When their prenylation is inhibited by statins, these proteins become less active. This leads to a cascade of downstream changes that are often called pleiotropic effects—actions unrelated (or only partly related) to circulating LDL levels.
These effects are well-established in lab, animal, and human studies and include:
Boosting endothelial nitric oxide production (improving blood vessel relaxation and function)
Reducing vascular inflammation (e.g., lowering C-reactive protein [CRP], as dramatically shown in the JUPITER trial)
Stabilizing atherosclerotic plaques
Antioxidant activity
And—perhaps most relevant here—antithrombotic and anticoagulant actions
The antithrombotic effects operate at multiple levels of the clotting system and appear at least partly independent of LDL lowering.
Statins downregulate tissue factor (TF) expression in endothelial cells and macrophages. TF is the primary initiator of the extrinsic coagulation pathway—when a plaque ruptures, TF exposure triggers rapid thrombin generation and clot formation. By inhibiting Rho/ROCK signaling (a pathway activated when Rho is prenylated), statins suppress TF. They also upregulate thrombomodulin on endothelial cells; thrombomodulin shifts thrombin’s activity toward activating protein C, a natural anticoagulant that inhibits clotting factors.
On platelets, statins reduce thromboxane A2 production, impair activation and aggregation, improve fibrin clot structure (making clots easier to break down), and enhance fibrinolysis by lowering plasminogen activator inhibitor-1 (PAI-1).These mechanisms kick in quickly—often within weeks—and many are mediated directly through the mevalonate pathway inhibition, not just LDL particle removal.
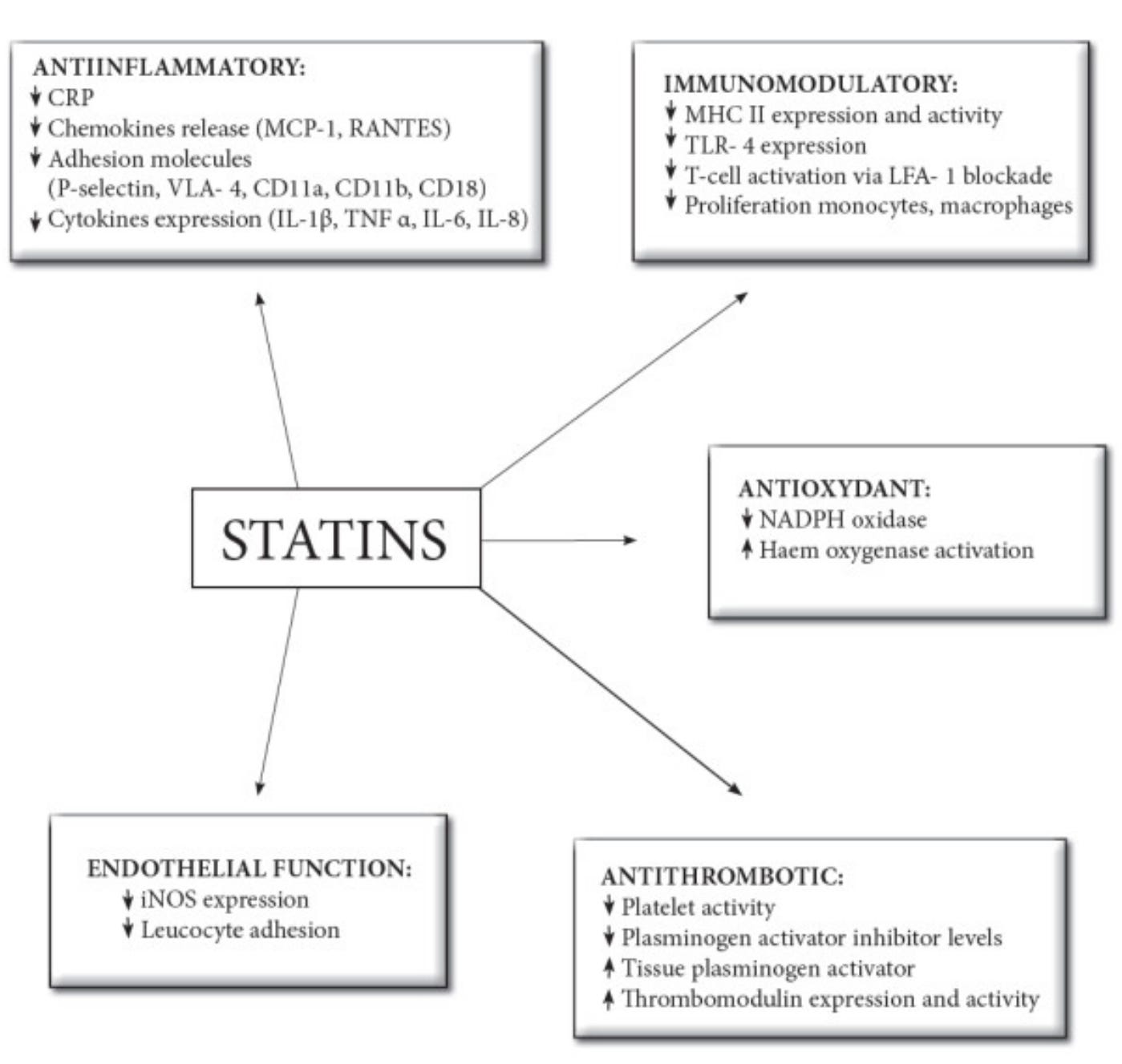
Supporting evidence that these antithrombotic effects aren’t solely from cholesterol lowering includes:
In the landmark JUPITER trial (rosuvastatin in people with normal LDL but high CRP), rosuvastatin reduced venous thromboembolism (VTE) by 43% (hazard ratio 0.57; 95% CI 0.37–0.86). VTE (deep vein thrombosis or pulmonary embolism) has only a weak link to high cholesterol at best—unlike arterial events—so this benefit is widely attributed to pleiotropic, non-LDL effects like reduced TF expression or improved fibrinolysis. (2)
Head-to-head comparisons sometimes favor statins over pure LDL-lowering agents like ezetimibe for certain non-lipid outcomes. For example, higher-dose atorvastatin reduced platelet activation markers (e.g., P-selectin) more than low-dose atorvastatin plus ezetimibe, despite similar LDL drops—suggesting statin-specific effects beyond cholesterol reduction. (3)
Reviews of experimental and clinical data consistently highlight that statins exert anticoagulant effects via Rho/ROCK inhibition, TF downregulation, thrombomodulin upregulation, and platelet modulation—often independent of LDL changes. (4)
In short, when a vulnerable plaque ruptures or erodes, the immediate danger is acute thrombosis—the clot that blocks the artery. By stabilizing plaques (via anti-inflammatory actions) and directly blunting the clotting response (via TF suppression, thrombomodulin boost, and antiplatelet effects), statins may prevent events faster and more effectively than LDL reduction alone would predict. This could explain why benefits sometimes appear early in trials (months, not years) and why greater LDL drops don’t always translate to proportionally greater mortality benefits. Mechanistically, the antithrombotic pleiotropy is a major contributor within statin-treated patients.
What This Means for Patients and Practice
If the dominant benefit in many people is coming from anti-inflammatory + antithrombotic pleiotropy rather than pure LDL reduction, several implications follow:
1. Mortality messaging needs honesty. In primary prevention, telling healthy people “this will help you live longer” overstates the data for most. Absolute risk reductions are small; number-needed-to-treat for all-cause mortality is often in the hundreds over 5 years.
2. LDL targets may not tell the whole story. Some patients achieve massive LDL drops but see limited clinical gain; others with modest drops do better. Inflammation and thrombotic tendency (measurable via hs-CRP, fibrinogen, or even simple platelet function in research settings) might matter more than we acknowledge.
3. Mechanism matters for the future. If antithrombotic pleiotropy is a major driver, we should study whether low-dose statins + other targeted anti-coagulants/anti-inflammatories (or newer agents hitting Rho or TF pathways) could achieve similar protection with fewer side effects. We should also ask why PCSK9 inhibitors (pure LDL lowering, no mevalonate inhibition) sometimes show slightly different patterns in inflammation or certain subgroups.
4. Shared decision-making. Every patient deserves to know the absolute benefits, the lack of consistent mortality reduction in primary prevention, the pleiotropic story, and the side-effect profile (myalgia, new-onset diabetes risk ~10–20% relative increase, etc.). Lifestyle—diet, exercise, smoking cessation, sleep, stress—still moves the needle on inflammation and clotting far more powerfully than any pill for most low-to-moderate risk people.
The statin story isn’t black-and-white and the widespread, long-term use in millions of lower-risk adults rests on a narrative that emphasizes LDL numbers while downplaying the mortality data and the mechanistic complexity.
It’s time for a more nuanced conversation: one that acknowledges the pleiotropic reality, especially the antithrombotic effects, and stops pretending cholesterol is the only—or even the main—actor. Until we design trials that better disentangle these mechanisms (e.g., statin vs. pure LDL-lowering agent head-to-head with thrombosis biomarkers), we should prescribe with humility, not hype, marketing and dogma.
What do you think—have you or a loved one been told statins are essential purely for cholesterol, without this fuller picture?
Coming Next: Statins and the GLP-1 Paradox—Are Cholesterol Pills Quietly Undermining Our Natural Appetite and Blood Sugar Control?
We’ve already unpacked how statins often fail to deliver clear all-cause mortality wins in primary prevention and how their real magic might lie in antithrombotic/anti-inflammatory pleiotropy rather than just LDL drops. But what if these same drugs are quietly sabotaging another key metabolic player?
Emerging evidence suggests statins can reduce circulating levels of glucagon-like peptide-1 (GLP-1)—the very hormone boosted by blockbuster drugs like Ozempic, Wegovy, Mounjaro, and others that millions now take for diabetes, obesity, and cardiometabolic protection. GLP-1 naturally enhances insulin secretion, slows gastric emptying, curbs appetite, and promotes satiety. When levels drop, it could theoretically worsen insulin resistance, glucose control, and even contribute to the modest diabetes risk long associated with statins.
A high-profile 2024 study in Cell Metabolism found that atorvastatin treatment led to a sharp decline in blood GLP-1 (nearly 50% in some human data points), linked to gut microbiota shifts that impair bile acid metabolism (specifically reduced ursodeoxycholic acid/UDCA, a key stimulator of GLP-1 secretion from intestinal L-cells). This microbiota-dependent mechanism aggravated insulin resistance in models, raising questions about long-term metabolic trade-offs. (5)
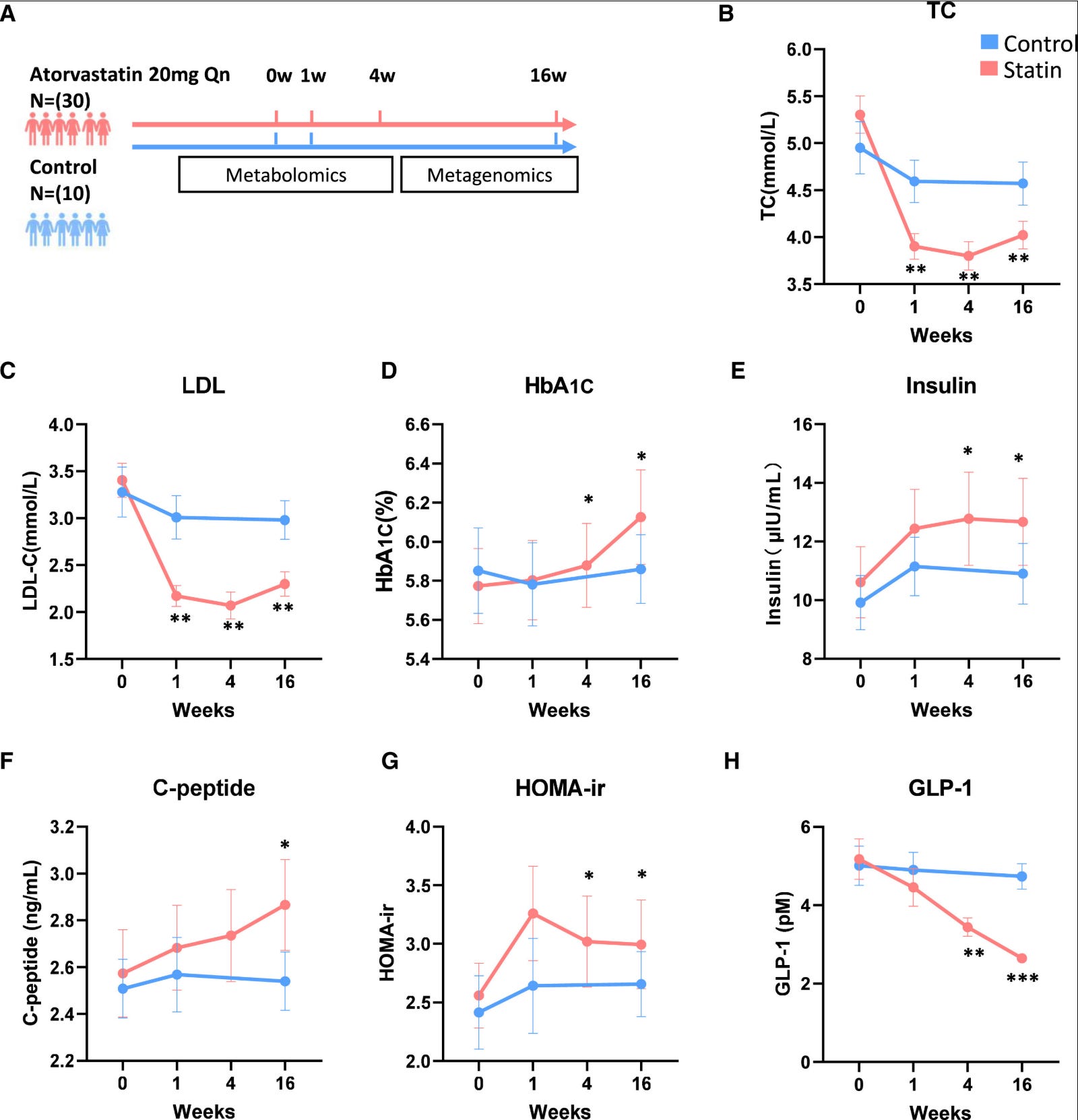
Supporting mechanistic context comes from related work on statin effects in beta cells and incretin pathways (e.g., simvastatin impairing GLP-1 potentiated insulin secretion in cell lines), but the She et al. paper is the most direct human-relevant hit on endogenous GLP-1 reduction.
In a world where cardiologists prescribe statins to prevent heart events and endocrinologists/endocrinologists add GLP-1 agonists to fight metabolic syndrome, could one drug be quietly counteracting the benefits of the other? Or is the effect too small to matter clinically? We’ll dive into the data, the microbiome angle, potential fixes (like bile acid modulation), and what this means for patients on both classes.
Stay tuned—drop a comment if you’d like me to cover anything else and share share share…Your experiences with statins + GLP-1 meds are especially welcome.
References
1. Ray KK, Seshasai SR, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med. 2010;170(12):1024-1031. doi:10.1001/archinternmed.2010.182
2. Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360(18):1851-1861. doi:10.1056/NEJMoa0900241
3. Piorkowski M, Fischer S, Stellbaum C, et al. Treatment with ezetimibe plus low-dose atorvastatin compared with higher-dose atorvastatin alone: is sufficient cholesterol-lowering enough to inhibit platelets?. J Am Coll Cardiol. 2007;49(10):1035-1042. doi:10.1016/j.jacc.2006.10.064
4. Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res. 2017;120(1):229-243. doi:10.1161/CIRCRESAHA.116.308537
5. She J, Tuerhongjiang G, Guo M, et al. Statins aggravate insulin resistance through reduced blood glucagon-like peptide-1 levels in a microbiota-dependent manner. Cell Metab. 2024;36(2):408-421.e5. doi:10.1016/j.cmet.2023.12.027
I’m going to pass this along to my Dr. (in the VA medical system). She wanted to subscribe me statins. I politely declined and because all my other lab markers were very good, she agreed.
Thanks J!! I like to take control of my own health and am guided by you and a few others that back up their research. The more I learn the more confident I am in all aspects of life.